
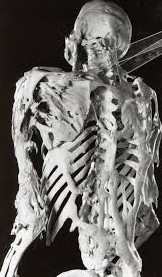
La fibrodysplasie ossifiante progressive (FOP)
La fibrodysplasie ossifiante progressive (FOP), autrement appelée la maladie de l’homme de pierre, est une maladie héréditaire sévèrement invalidante du tissu conjonctif, caractérisée par des malformations congénitales des gros orteils et une ossification hétérotopique progressive formant de l'os qualitativement normal dans des tissus extrasquelettiques.
Une évolution par poussées
La fibrodysplasie ossifiante progressive se caractérise par le développement d’ossifications dites « hétérotopiques », c’est-à-dire localisées à des endroits du corps où elles ne devraient pas se trouver : dans des muscles squelettiques et des tissus conjonctifs (ligaments, tendons, capsules articulaires...). Ce néo-tissu osseux est une zone plus dure que les tissus environnants et souvent douloureuse.
Les ossifications hétérotopiques sont en général précédées par une inflammation locale (gonflement, rougeur et chaleur de la peau, douleur) et elles peuvent s’accompagner de fièvre. Leur survenue se fait par poussées, spontanées ou à la suite d’une lésion musculaire (étirement, choc, injection intramusculaire...) ou d’une infection virale comme la grippe.
À noter que toutes les poussées d’inflammation locale ne sont pas suivies de l’apparition d’une ossification hétérotopique. Par ailleurs, ossification hétérotopique et inflammation locale peuvent survenir à deux endroits différents.
Des complications possibles pour d’autres organes
La FOP peut entrainer :
- un déficit de l’audition
des difficultés d’alimentation et de déglutition, par ossification de l’articulation de la mâchoire et des muscles masticateurs,
des troubles de conduction et du rythme cardiaque,
une insuffisance respiratoire, en raison de l’ossification de muscles respiratoires et de déformations de la colonne vertébrale,
une phlébite et une embolie pulmonaire, en raison du manque de mouvements.
Quelle est la cause de la FOP ?
La fibrodysplasie ossifiante progressive est une maladie génétique. Elle résulte d’un variant pathogène (mutation), quasiment toujours identique, au niveau du gène ACVR1 également appelé ALK2. Le diagnostic de la maladie repose sur un test génétique, pratiquée à partir d’une simple prise de sang.
Identifié en 2006, le gène code une protéine (ACVR1/ALK2) impliquée dans le développement des os : le récepteur de type I de l’activine A. Les mutations responsables de la FOP entrainent l’hyperactivation du gène ACVR1. On parle de mutation « gain de fonction », avec pour conséquence le développement excessif de tissu osseux, à des endroits inhabituels.
Une mutation le souvent non héritée
La FOP se transmet sur le mode autosomique dominant. Nous possédons tous deux exemplaires du gène ACVR1. Il suffit qu'une anomalie touche un seul de ces deux exemplaires pour développer la maladie. Cette anomalie peut être transmise (risque de 50%) par l’un des parents, lui-même malade.
En pratique cependant, rares sont les cas de FOP transmis par un parent atteint. La plupart de cas sont au contraire « sporadiques » : la mutation responsable de la maladie survient spontanément, chez un seul enfant, sans autre cas de FOP dans la famille. Et la probabilité que les parents, non atteints, aient un deuxième enfant atteint de FOP est infime (1%). Elle est liée à la possibilité d’une « mosaïque germinale » : un clone d’ovules ou de spermatozoïdes est porteur de la mutation, et les autres ovules ou spermatozoïdes n’en sont pas porteurs.
Quels symptômes ?
Bien que les patients naissent avec la maladie, elle est d’abord asymptomatique. Seule une malformation des gros orteils est présente : ils sont déviés vers l’intérieur et plus courts.
Dès les 10 premières années, des tuméfactions douloureuses surviennent par poussées. Elles sont souvent favorisées par une injection intramusculaire, une infection virale, une blessure, une chute, un stress, un étirement musculaire ou de la fatigue. Ainsi, en cas de suspicion de la maladie, tout geste invasif est contre-indiqué, sous peine de déclencher une poussée.
Actuellement, l’espérance de vie de la maladie de l’homme de pierre est de 40 ans. La majorité des patients nécessitent un fauteuil roulant dans la vingtaine. Le décès survient par insuffisance respiratoire.