
Article
Syndrome de Lynch
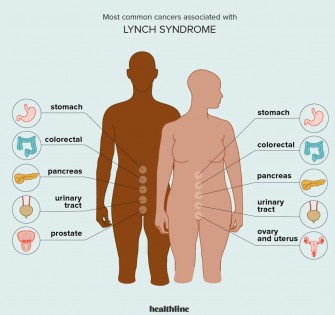
Le syndrome de Lynch est une maladie génétique, caractérisée par une augmentation du risque de développer certains cancers, en particulier le cancer colorectal. Il serait impliqué dans 2 à 3 % des cancers colorectaux. Le mode de transmission est autosomique dominant.
Le syndrome de Lynch est causé par une mutation d’un gène impliqué dans la réparation de l’ADN. L’ADN est le support de l’information génétique de chaque cellule. Il peut subir des erreurs lors de sa duplication ou de son exposition à des agents mutagènes (rayons UV, tabac, etc.). Normalement, ces erreurs sont corrigées par des mécanismes de réparation de l’ADN. Mais lorsque l’un de ces mécanismes est défectueux, les erreurs s’accumulent et peuvent entraîner une transformation des cellules en cellules cancéreuses. Le syndrome de Lynch est une maladie autosomique dominante, ce qui signifie qu’il suffit d’hériter d’une copie mutée du gène pour être atteint. Le gène muté peut être transmis à la descendance par le père ou la mère, avec un risque d’environ 50 %. Il existe plusieurs gènes impliqués dans le syndrome de Lynch, notamment MLH1, MSH2, MSH6, PMS2 et EPCAM. La mutation de l’un de ces gènes entraîne une instabilité des microsatellites (IMS), qui sont des séquences répétées de l’ADN. L’IMS est un marqueur biologique du syndrome de Lynch.
Le syndrome de Lynch entraîne-t-il des symptômes ?
Le syndrome de Lynch ne provoque pas de symptômes spécifiques, mais il augmente le risque de développer certains cancers, en particulier le cancer du côlon et de l’endomètre. Ces cancers peuvent se manifester par des signes tels que :
- La présence de sang dans les urines ou les selles
- Des douleurs abdomino-pelviennes
- Des troubles du transit (diarrhée, constipation, ou alternance des deux)
- Une perte de poids inexpliquée
- Une fatigue persistante
- Des saignements vaginaux anormaux
- Une masse palpable au niveau du ventre ou du pelvis
Le syndrome de Lynch peut être associé à d’autres troubles, comme des polypes du côlon, des kystes ovariens, des tumeurs bénignes de la peau, des anomalies dentaires… Les cancers liés au syndrome de Lynch ont tendance à apparaître plus tôt que les cancers non héréditaires.
L’âge moyen de diagnostic est de 45 ans pour le cancer du côlon et de 50 ans pour le cancer de l’endomètre. Le risque de développer un cancer du côlon au cours de la vie est de 50 à 80 % chez les personnes atteintes du syndrome de Lynch, bien plus élevé que chez les autres patients. Il en est de même pour le cancer de l’endomètre, avec un risque estimé entre 25 et 60 %, contre seulement 3 % dans la population générale.
Diagnostic du syndrome de Lynch
La recherche du syndrome de Lynch repose sur une anamnèse cherchant à identifier les antécédents médicaux et familiaux évocateurs d’une prédisposition génétique au développement de pathologies cancéreuses. Bien souvent, le syndrome de Lynch est diagnostiqué après le développement du cancer dont il a favorisé l’apparition. Le diagnostic repose sur l’imagerie médicale, qui permet de localiser la tumeur cancéreuse, et surtout sur la biopsie, qui permet de prélever des cellules tumorales afin de les analyser en laboratoire. L’analyse en laboratoire des cellules cancéreuses prélevées peut permettre d’établir le profil génétique de la tumeur, ainsi que de déterminer son stade, son grade et ses caractéristiques moléculaires afin d’élaborer le protocole de traitement le mieux adapté. Lorsqu’un syndrome de Lynch est diagnostiqué, l’équipe médicale peut proposer une consultation d’oncogénétique aux apparentés du patient s’ils le souhaitent, et mettre en place un protocole de surveillance.
Surveillance du syndrome de Lynch
Les personnes atteintes du syndrome de Lynch doivent bénéficier d’une surveillance régulière pour détecter précocement les éventuels cancers et les traiter efficacement. La surveillance repose essentiellement sur :
- Une coloscopie totale tous les 2 ans
- Une gastroscopie tous les 2 à 4 ans selon l’histoire familiale
- Une échographie endovaginale chez la femme
- Une hystéroscopie s’il existe une prolifération anormale du tissu de l’endomètre
Le suivi est personnalisé en fonction du type de mutation, du type de cancer familial et des facteurs de risque du patient. Il peut inclure d’autres examens, comme l’analyse d’urine ou l’examen dermatologique.