

Amaurose congénitale de Leber
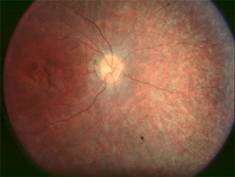
Amaurose de Leber due à des mutations de RPE65
(Christian Hamel, Inserm Montpellier France)
En 1869, Théodor Leber, ophtalmologiste allemand, décrit pour la première fois une maladie rétinienne survenant dès la naissance qu'il appelle "rétinite pigmentaire in utero" laissant ainsi son nom à une dystrophie rétinienne gravissime débutant en période anténatale. C’est une forme de rétinite pigmentaire entraînant l'apparition d'une quasi-cécité dès l'enfance. Elle est due à des dysfonctionnements dans les communications entre les cellules rétiniennes photoréceptrices et celles de l'épithélium rétinien, la couche la plus profonde de la rétine, où le signal lumineux est converti en signal électrique, seul interprétable par le cerveau. L’amaurose (du grec amauroô : j’obscurcis) congénitale de Leber se définit donc comme la perte complète de la vue, due à une lésion de la rétine ou des voies optiques (neuropathie optique héréditaire de Leber.). Cette cécité est soit transitoire soit définitive. Il existe plusieurs formes d'amaurose. L'amaurose congénitale de Leber (LCA), appelée également amaurose congénitale tapétorétinienne de Leber, entraîne une altération complète d'origine congénitale de la rétine et une perte de totale et définitive de la vision. L'acuité visuelle existe néanmoins parfois au début de la vie, mais elle est particulièrement faible. Cette variété d'amaurose s'accompagne d'un nystagmus (mouvements incessants et saccadés de l'oeil).
Etiologie
Fréquence : Elle représente au moins 5% des dystrophies rétiniennes et constitue l’une des principales causes de cécité chez l’enfant.
C'est une maladie héréditaire transmise sur le mode autosomique récessif chez la plupart des patients mais quelques cas d'hérédité autosomique dominante ont été rapportés.
La LCA regroupe un ensemble de dystrophies rétiniennes dont l’origine génétique est très hétérogène et dont la classification est encore incomplète. Les premiers travaux (J. Kaplan) de démembrement génétique ont abouti en 1996 à l'identification d'un gène codant une enzyme clé de la cascade de transduction visuelle puisqu'elle permet le retour à l'état d'obscurité après stimulation rétinienne par la lumière. Les altérations de ce gène conduisent donc à une impossibilité pour la rétine de retrouver son état d'obscurité, ce qui équivaut fonctionnellement à une rétine illuminée en permanence. Il n'est donc pas surprenant d'observer chez les nouveau-nés atteints une aversion pour la lumière et un handicap visuel gravissime. L'année suivante, en 1997, une équipe allemande ( Andreas Gal ) rapporta les mutations d'un gène de l'épithélium pigmentaire de la rétine chez des patients atteints de rétinite pigmentaire très précoce de l'enfance. Plusieurs autres mutations touchant des gènes différents peuvent être responsables de l’amaurose congénitale de Leber. A ce jour, les chercheurs ont identifié huit gènes. Parmi eux, le gène RPE65 qui code pour une protéine spécifiquement exprimée dans l’épithélium rétinien. Cette protéine a récemment été identifiée comme étant une enzyme qui recycle une protéine permettant la synthèse du pigment indispensable à la vision : le chromophore 11-cis retinal.
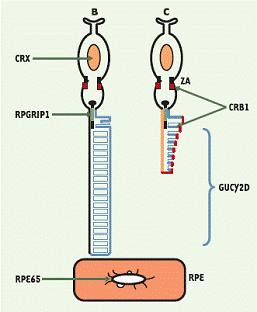
Schéma des photoneurones avec les localisations subcellulaires
des protéines codées par les gènes LCA
On estime que 10 % des enfants atteints par l'amaurose congénitale de Leber sont porteurs d'une mutation du gène RPE65. Le gène RPE65 code une protéine du même nom qui intervient dans la synthèse du pigment visuel (rhodopsine). Les patients ayant des mutations dans ce gène ont donc un déficit dans la synthèse du pigment visuel, et par conséquent ont des photorécepteurs qui réagissent très peu à la lumière. Il a été montré que chez la souris déficiente en RPE65, l'apport d'une forme artificielle d'un des composants de ce pigment visuel (le 9-cis-rétinal), permet de retrouver de la vision. Malheureusement ceci ne peut être envisagé chez l'homme pour l'instant car ce composant s'accumulerait et risquerait d'être préjudiciable à la rétine.
Il existe ainsi dans l’amaurose de Liber un extrême plurigénisme (hétérogénéité génétique). Sur les huit gènes différents reconnus à l’heure actuelle, les mutations de trois d'entre eux sont responsables de la maladie du premier groupe de patients (31 % des cas). Les mutations des cinq autres gènes concernent la maladie du second groupe (21% des cas). Il existe par ailleurs une forte corrélation entre l'histoire naturelle de la maladie et le gène en cause, permettant ainsi de sélectionner les gènes à étudier en priorité chez un nouveau patient. C'est la raison pour laquelle l'anamnèse et les tout premiers symptômes observés chez le nouveau né sont de la plus grande importance.
Diagnostic
L’amaurose congénitale (LCA) est une entité clinique caractérisée par un déficit visuel, diagnostiqué à la naissance ou dans les premiers mois de la vie.
Les enfants atteints présentent des difficultés à fixer et à suivre du regard car ils n'ont pas ou très peu de sensibilité rétinienne aux stimuli visuels.
L'activité électrorétinographique est profondément altérée ou inexistante. L'aspect du fond d'oeil est souvent normal dans les premiers mois de la vie mais évolue vers une atrophie chorio-rétinienne irréversible avec des remaniements pigmentaires. On trouve parfois dans la région maculaire une lésion à l'emporte-pièce. Les patients présentent un nystagmus et enfoncent souvent les doigts dans leurs yeux.
Le fond d'oeil décèle une pigmentation (coloration) anormale. L'électrorétinogramme (enregistrement de la réponse électrique à une excitation lumineuse au niveau de la rétine) est plat. Cataracte (opacité du cristallin) Kératocône : déformation progressive de la cornée en forme de cône, liée à un amincissement de celle-ci par suite d'une anomalie de son collagène (protéine fibreuse participant à la constitution de la trame de soutien des tissus de l'organisme).
D’un point de vue physiologique, la maladie s’explique par une mauvaise communication entre les cellules réceptrices de la lumière (les photorécepteurs) et l’épithélium pigmentaire rétinien (EPR). L’épithélium pigmentaire rétinien est la couche la plus profonde de la rétine, qui se trouve en contact étroit avec les photorécepteurs. L’EPR accomplit des tâches essentielles pour la vie et le fonctionnement des photorécepteurs. Il recycle et transforme notamment les molécules impliquées dans la conversion du signal lumineux en signal électrique, seul signal interprétable par le cerveau.
Dans cette forme anténatale et gravissime la maladie se manifeste en période néonatale par une atteinte totale de la fonction des cônes de la rétine. Pour cette raison, les nourrissons ont un comportement de non voyant qu'ils manifestent par une absence de poursuite oculaire, un nystagmus congénital, une intolérance sévère à la lumière et une acuité visuelle nulle. Cette forme correspond à la description de Théodor Leber et les patients peuvent être considérés comme souffrant d'une maladie des cônes à début anténatal entraînant une cécité qui n'évoluera pas tout au long de la vie.
A coté de cette dystrophie rétinienne congénitale ont étaient décrites des rétinites pigmentaires très précoces de l'enfance, débutant vers l'âge de deux ans. Néanmoins, certains symptômes étaient communs avec les manifestations précoces de l'amaurose congénitale de Leber comme le nystagmus congénital et la réduction sévère de l'acuité visuelle.
Les patients de ce deuxième groupe peuvent être considérés atteints d'une maladie à début post-natal correspondant à l'extrémité d'un spectre de gravité des rétinites pigmentaires. Dans ce cas, la maladie sera évolutive : une amélioration de la fonction visuelle sera notée dans les premières années de vie puisque le petit enfant aura, certes, une acuité visuelle faible mais mesurable qui lui permettra d'avoir non seulement des souvenirs visuels mais également un début de scolarité «en noir». Malheureusement, dès la deuxième décennie de vie, la maladie va s'aggraver pour aboutir à un tableau de cécité légale à la fin de la deuxième décennie.
Thérapie génique
Alors qu'aucun traitement curatif n’existait pour cette maladie génétique, des résultats encourageants ont été obtenus depuis 2001 en thérapie génique dans un modèle canin de cette maladie et depuis l’année 2007, chez l’homme.
En effet, en 2001, G M Acland et col. ont décrit une thérapie génique de chiens présentant une affection comparable à l'amaurose congénitale de Leber, avec succès. Les auteurs ont utilisé un adeno-associated virus (AAV) pour transmettre le gène normal RPE65 aux animaux malades qui ont recouvré une certaine vision.
Les chiens homozygotes présentant une mutation des 2 copies de RPE65 vont devenir progressivement aveugles par dégénérescence des photorécepteurs. In vitro l'équipe de G M Acland a combiné le gène RPE65 normal à un virus AAV, puis a transmis le virus combiné dans la culture de cellules déficientes. L'immunohistochimie a permis de mettre en évidence l'apparition du gène normal dans les cellules malades. In vivo, les auteurs ont administré le virus combiné à trois chiens et ont surveillé l'électrorétinogramme (ERG) pour étudier le fonctionnement de la rétine. L'injection s'est faite sous la rétine au niveau de l'oeil droit et dans le vitré au niveau de l'oeil gauche.
L'ERG des chiens a mis en évidence une augmentation du tracé et donc un fonctionnement rétinien amélioré de l'oeil droit. L'ERG de l'oeil gauche n'a pas montré d'amélioration. L'étude a été faite également avec un pupillomètre et on a constaté une meilleure réponse pour les yeux droits traités en injection sous-rétinienne. Le comportement des chiens a été étudié également et on s'est rendu compte que les chiens traités évitaient les obstacles situés sur leur côté droit; les chiens non traités n'évitaient aucun obstacle. A 99 jours de l'injection, les yeux ont été étudiés sur le plan histochimique. La PCR a permis de se rendre compte que la rétine exprimait bien le gène sain.
En 2006, une équipe de chercheurs français a également réussi à rendre la vue à 7 sur 8 chiens touchés par cette affection génétique de la rétine. L’équipe nantaise de F. Rolling, a utilisé une technique de thérapie génique semblable à celle de G M Acland : Elle a recouru à des virus de type AAV, qui ciblent les cellules de la rétine à l’origine du dysfonctionnement visuel.Techniquement le traitement a été appliqué à un seul œil pour chaque chien. Chez 7 des 8 animaux, les chercheurs ont pu observer le retour d’une activité électrique au niveau de la rétine, ce qui témoigne de la restauration de la fonction des photorécepteurs. Cette équipe nantaise prévoit un premier essai humain sur des patients âgés de 8 à 18 ans dans les années à venir.
En mai 2007, une équipe de médecins et de biologistes britannique, dirigée par le professeur Robin Ali (University College, Londres), a annoncé avoir réalisé l'injection au sein de la rétine - via l'humeur vitrée – d’un gène (RPE65) chez une personne de 23 ans , atteinte par l'amaurose congénitale de Leber. Dans cette expérience, le gène normal est véhiculé par un virus vecteur membre de la famille des adeno-associated virus (AAV). L’opération s’est déroulée à l'hôpital ophtalmologique Moorfields de Londres. Elle devrait être reproduite sur une douzaine de personnes lors d'un essai clinique de phase I. Les premiers résultats sont attendus dans les mois à venir.
Bibliographie
G M Acland, G D Aguirre, J Ray, Q Zhang, T S Aleman, A V Cideciyan, S E Pearce-Kelling, V Anand, Y Zeng, A M Maguire, S G Jacobson, W W Hauswirth & J Bennett. Gene therapy restores vision in a canine model of childhood blindness. Nature Genetics Volume 28 May 2001; 92 - 95.
Agurirre GD, Baldwin V, Pearcez-Kelling S, et al. Congenital stationary night blindness in the dog: common mutation in the RPE65 gene indicates founder effect. Mol Vision 1998; 4: 23.
Cremers FPM, van den Hurk JAJM, den Hollander AI. Molecular genetics of Leber congenital amaurosis. Hum Mol Genet 2002; 11: 1169-76.
S. Gilgenkrantz L’opsine en liberté, une des causes de l’amaurose congénitale de Leber . M/S n° 3, vol. 20, 274 mars 2004
Hamel C, Marlhens F. Des mutations de gènes contrôlant le métabolisme des rétinoïdes 11-cis responsables de dystrophies rétiniennes sévères. Med Sci (Paris) 1998; 14: 754-7.
Kaplan J, Rozet J, Gerber S, et al. Des gènes pour les dystrophies rétiniennes des enfants. Med Sci (Paris)1995; 11: 325-35.
J. Kaplan. Le point sur l'amaurose congénitale de Leber. Le Rétino n° 52, décembre 2004
G Le Meur, K Stieger, A J Smith, M Weber, J Y Deschamps, D Nivard, A Mendes-Madeira, N Provost, Y Péréon, Y Cherel, R R Ali, C Hamel, P Moullier and F Rolling. Restoration of vision in RPE65-deficient Briard dogs using an AAV serotype 4 vector that specifically targets the retinal pigmented epithelium . Gene Therapy advance online publication, October 5, 2006.
Perrault I, Rozet JM, Calvas P, et al. Retinal specific guanylate cyclan gene mutations in Leber’s congenital amaurosis. Nat Genet 1996; 14: 461-4.
Remé CE, Wenzel A. The dangers of seeing light in the dark. Nat Genet2003; 35: 115-6.
Wooruff ML, Wang Z, Chung HY, et al. Spontaneous activity of opsin apoprotein is a cause of Leber congenital amaurosis. Nat Genet2003; 35: 158-64.