
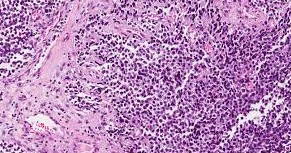
Le rhabdomyosarcome
Le rhabdomyosarcome (RMS) est un cancer qui prend naissance dans les cellules musculaires ou dans des formes précoces des cellules musculaires. La tumeur cancéreuse (maligne) est un groupe de cellules cancéreuses qui peuvent envahir les tissus voisins et les détruire. La tumeur peut également se propager (métastases) à d’autres parties du corps. Le RMS affecte surtout les enfants. Un muscle est une sorte de tissu mou. Les différents types de tissus mous accomplissent diverses fonctions. Ils donnent sa forme au corps et lui servent de soutien. Ils protègent aussi d’autres tissus, les os et les organes, en les maintenant ensemble. On trouve de nombreux types différents de tissus mous dans tout le corps, comme la graisse, les muscles, les tendons, les ligaments et les vaisseaux sanguins.
Le RMS peut prendre naissance dans n’importe quel muscle. Les régions du corps les plus affectées sont celles-ci :
- tête et cou (comme près d’un œil, au fond du nez, dans la gorge, près de la colonne vertébrale au niveau du cou ou dans le tissu entourant le cerveau);
- appareil génito-urinaire (comme la vessie, la vulve, le vagin, l’utérus, les ovaires, la prostate et autour des testicules);
- extrémités (comme un bras, une jambe, une main ou un pied);
- tronc (comme le thorax, l’abdomen, le bassin ou le diaphragme).
Le type le plus fréquent de RMS est le rhabdomyosarcome embryonnaire. Il représente de 50 à 70 % de tous les RMS chez l’enfant. Le RMS est le type le plus courant de sarcome des tissus mous chez l’enfant. Il apparaît rarement chez l’adulte.
La cause de la plupart des cas de rhabdomyosarcome est inconnue. Dans de rares cas, le rhabdomyosarcome a été associé à une mutation génétique qui affecte la formation de cellules au cours du développement fœtal. Les scientifiques continuent d'explorer la cause de ce sarcome des tissus mous.
Comment se développe un rhabdomyosarcome ?
La formation d’un rhabdomyosarcome, comme tous les cancers, résulte d’une modification de l’ADN et plus précisément d’un gène (ou portion de cet ADN). En cas de mutation génétique, les cellules des tissus mous (principalement les muscles et les tissus de soutien des organes) ne se comportent pas normalement, ce qui prédispose au développement du rhabdomyosarcome.
Au début de la maladie, les cellules cancéreuses sont peu nombreuses et localisées. Par la suite, le rhabdomyosarcome peut s’étendre aux ganglions avoisinants (surtout lorsqu’il touche les membres). En l’absence de traitement, des cellules cancéreuses se détachent de la tumeur, empruntent les vaisseaux lymphatiques et/ou sanguins et s’implantent dans d’autres parties du corps comme les ganglions lymphatiques de l’abdomen et certains organes, notamment les poumons ou la moelle épinière... Les nouvelles tumeurs formées s’appellent des métastases.
Quels sont les facteurs de risque du rhabdomyosarcome ?
La cause du rhabdomyosarcome reste inconnue. Il existe néanmoins certains facteurs de risque hypothétiques identifiés.
Des facteurs génétiques
Certains syndromes génétiques (et donc les altérations génétiques qu’ils traduisent) peuvent faire augmenter le risque d’apparition d’un rhabdomyosarcome : le syndrome de Li-Fraumeni, la neurofibromatose de type 1, le syndrome de Beckwith-Wiedemann, le syndrome de Costello, le syndrome de Noonan
Des facteurs environnementaux hypothétiques
Certains facteurs de risques restent hypothétiques et doivent encore faire l’objet d’études afin de confirmer leur implication dans le déclenchement du rhabdomyosarcome :
- Un poids élevé à la naissance ;
- une exposition aux drogues, comme le cannabis (marijuana) ou la cocaïne, avant la naissance ;
- une exposition à de faibles doses de radiation lors d’examens médicaux, comme la radiographie ou la tomodensitométrie, avant la naissance
Quels sont les traitements du rhabdomyosarcome ?
La prise en charge du rhabdomyosarcome consiste le plus souvent en l’association de plusieurs traitements : une intervention chirurgicale, une chimiothérapie et parfois une radiothérapie.
La chirurgie
Elle est réservée aux cas où elle peut être complète et sans conséquence fonctionnelle. Elle est donc proposée surtout pour les petites tumeurs aisément accessibles.
La chimiothérapie
Tous les enfants sont traités par chimiothérapie. Les médicaments les plus fréquemment utilisés sont la vincristine, la dactinomycine, le cyclophosphamide, la doxorubicine, l’ifosfamide et l’étoposide. Le topotécan et l’irinotécan sont d’autres médicaments pouvant également être utilisés.
La radiothérapie
Elle est normalement utilisée si certains cancers demeurent après intervention chirurgicale ou si le cancer est considéré comme un cancer à risque intermédiaire ou élevé. Une forme ultra-précise de radiothérapie, la proton-thérapie, est indiquée dans les tumeurs de la tête et de l’appareil génital, pour limiter au maximum l’irradiation des tissus sains environnants. Une fois en rémission les patients sont suivis pendant de nombreuses années afin de prévenir les récidives, mais également afin de prendre en charge toute éventuelle séquelle du cancer et de ses traitements.
.